



Beeline: Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data
This post was also shared on Aditya’s blog
The full spectrum of biological behaviors are orchestrated by the activities of gene products. A small number of key molecules can completely redefine a cell’s fate - from a stem cell “committing” to a tissue specific developmental pathway, or a healthy cell turning cancerous. How can we understand the key cellular events that define complex biological behavior?
One approach to answering this question has been to directly measure the activities of gene products. RNA sequencing (RNA-seq) is one such technology that enables us to measure hundreds or thousands of genes simultaneously in a biological sample. There are an estimated 200,000 published RNA-seq datasets in the last two decades (Lachmann, 2018): how do we make sense of these large and complex datasets? A variety of computational approaches have been proposed to derive insights into biological processes from RNA-seq data. One area of impact has been the elucidation of the genetic circuitry, i.e. the regulatory mechanisms underlying biological processes.
This “bulk” RNA-seq data comes with its limitations. First, in order to fully understand the changes in cellular states in a tissue we will need to sequence samples periodically, which can be expensive and time consuming. Moreover, extracting samples from whole tissues obfuscates the differences between the cells comprising the tissue, differences that might be vital in the developmental context of the tissue. Recent advances in sequencing technologies have enabled the investigation of gene expression at the single cell level. In contrast to bulk RNA-seq measurements, single cell RNA-seq (scRNA-seq) measurements offer snapshots of the heterogeneity present in a population of cells, which can also be used to recreate developmental timeline of cells.
Using scRNA-seq data to infer GRNs comes with a new set of challenges which has led an active development of computational techniques, with a dozen methods published since 2015. Unfortunately, an experimentalist seeking to analyze a new dataset faces a daunting task in selecting an appropriate inference method. Which method is appropriate for my dataset? How long might the method take to run? How can I test the accuracy of the predicted network? There is thus a pressing need for a systematic evaluation and benchmarking of GRN inference algorithms for single-cell gene expression data.
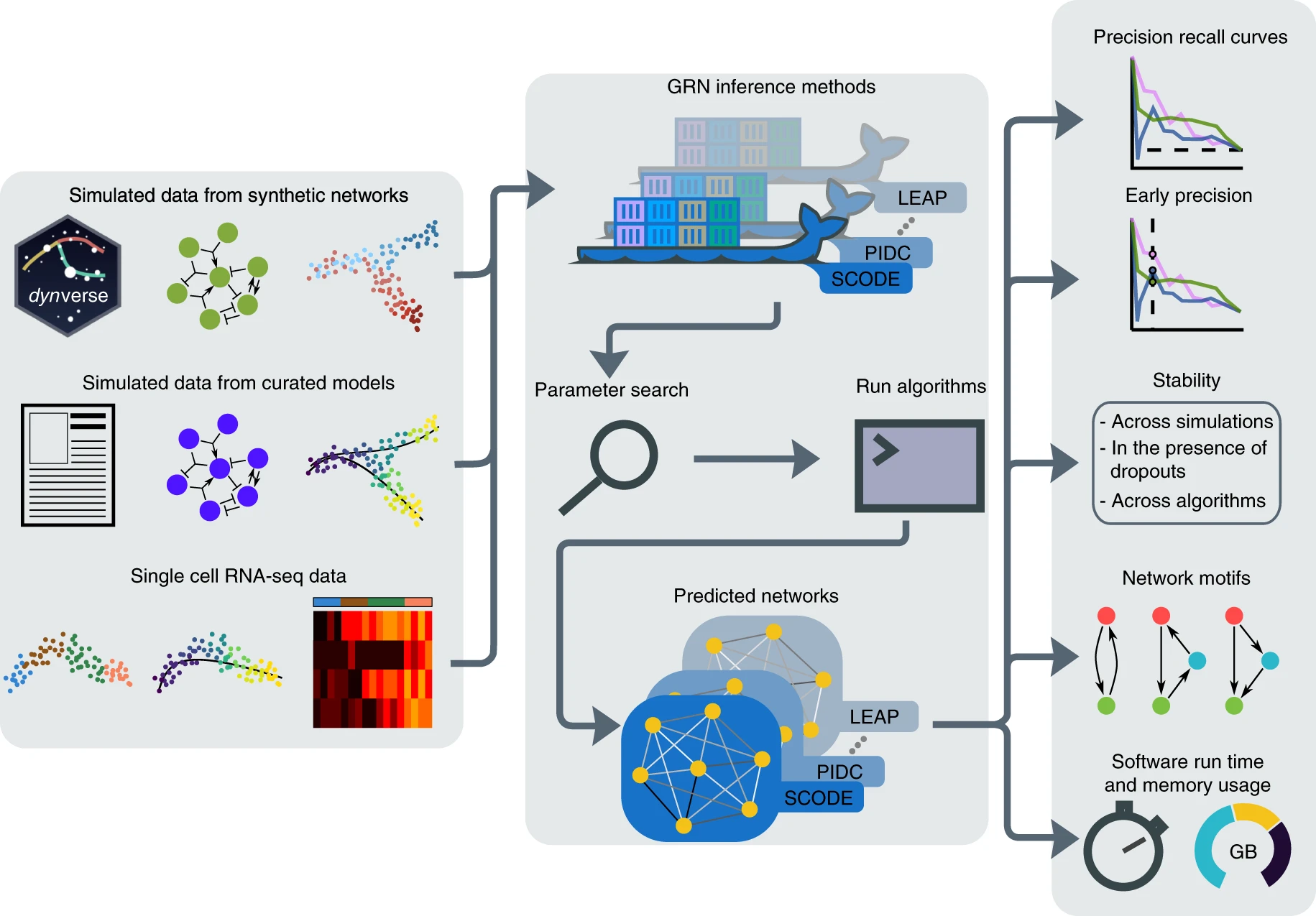
We developed BEELINE, a comprehensive evaluation framework to address this need. The first challenge we faced was that the methods were implemented across 5 different platforms. The solution we came up with draws from the industry standards for software virtualization. Using Docker, BEELINE provides a standardized interface to these different methods, lowering the barrier to future users of these methods. The next challenge was that there are no widely accepted ‘ground truth’ datasets, so we came up with a novel framework, BoolODE, to simulate single cell data from published models of tissue development. We also evaluated the GRN inference methods on experimental single cell datasets. Lastly, there was no standard tool to carry out a comprehensive evaluation of GRN inference methods (for either bulk or single cell RNAseq methods). BEELINE provides a consistent and extensible interface to evaluate methods in terms of accuracy, stability, and scalability.
While BEELINE identified several trends across the various evaluations, surprisingly the overall performance of the methods is less than ideal. One of the key insights was that the methods typically performed poorly on “branching” cellular trajectories, i.e. in developmental contexts leading to multiple terminal cell types starting from a stem cell stage. We noted that the reconstruction accuracy of the methods on our simulated data was reflective of that on experimental datasets. This demonstrates that our proposed framework can serve as a valuable tool to harness existing domain specific knowledge to create simulated datasets. Thus, BEELINE facilitates reproducible, rigorous and extensible evaluations of GRN inference methods for single-cell gene expression data. Large-scale projects such as the Human Cell Atlas and Tabula Muris will soon generate complex single-cell multi-omics data. The next generation of GRN inference algorithms will interrogate single cells along multiple modalities. We believe that scientists in this field will find BEELINE to be immensely valuable as they develop new approaches for GRN inference.
The BEELINE pipeline as well as the dockerized methods are available is available under an open source license at https://github.com/murali-group/beeline.